Sanidad negocia la adquisición del medicamento más caro del mundo: 2,47 millones por una única dosis
El Libmeldy puede curar por completo la leucodistrofia metacromática, una rara enfermedad genética mortal que frena el desarrollo de los niños al dañar las conexiones neuronales

El Ministerio de Sanidad negocia con la farmacéutica británica Orchard Therapeutics la financiación pública del Libmeldy, un innovador medicamento de una sola dosis indicado para el tratamiento de la leucodistrofia metacromática. Esta enfermedad genética muy rara detiene el desarrollo de los niños, la mayoría de las veces antes de que cumplan tres años, y es mortal al destruir las conexiones neuronales. El precio de salida del fármaco, 2,47 millones de euros, lo convierte en el más caro del mundo y deja muy atrás el Zolgensma de Novartis, indicado para la atrofia muscular espinal y disponible en la sanidad pública desde el pasado diciembre a un precio oficial de 1,95 millones de euros.
La buena efectividad demostrada en los ensayos por el Libmeldy y el elevado precio impuesto por Orchard Therapeutics aviva el debate sobre la escalada de precios de los nuevos medicamentos innovadores. “Estos precios llevan a una situación en la que lo éticamente aceptable y lo económicamente sostenible chocan. Y esto es algo muy complejo de gestionar. Si hay un tratamiento disponible que puede salvar vidas, hay que ofrecerlo. Pero esto tiene repercusiones. Cuando se dice que son fármacos destinados a un número pequeño de pacientes y que no dañan la sostenibilidad del sistema, se olvida que los nuevos tratamientos en desarrollo son cientos y que todos juntos sí suponen un reto gigantesco para la sanidad pública, que tiene unas necesidades enormes y unos recursos siempre limitados”, afirma Juan Oliva, profesor de Economía de la Salud en la Universidad de Castilla-La Mancha.
Cada año nacen en España entre tres y cinco niños con leucodistrofia metacromática, cuya incidencia estimada en Europa es de 1,1 casos por cada 100.000 nacimientos. “Es una enfermedad neurodegenerativa que se produce por mutaciones en el gen de la arisulfatasa A (ARSA) que dañan a la mielina, que es lo que recubre las conexiones neuronales. Son niños que tienen un buen desarrollo inicial, pero que un día empiezan a perder todas sus funciones de forma irreversible”, explica Mireia del Toro, jefa de la unidad de enfermedades metabólicas del Hospital Vall d’Hebron de Barcelona.
Los síntomas más frecuentes son alteraciones en el tono muscular —es habitual que los niños empiecen a tener problemas para caminar—, cambios súbitos en la personalidad y disminución de la función intelectual. El tratamiento funciona como un autotrasplante de células de médula ósea, en el que estas son modificadas en un laboratorio para que una vez devueltas al organismo sean capaces de producir la enzima que protege a la mielina.
“Es un gran paso adelante, un medicamento que según los resultados de los ensayos clínicos puede curar completamente con un buen perfil de seguridad, a niños que hasta ahora no tenían ninguna alternativa terapéutica”, afirma Del Toro.
Hasta el 60% de afectados por la enfermedad empiezan a desarrollar síntomas entre los seis y los 30 meses de vida, en la llamada “forma infantil tardía” que tiene una mortalidad del 75% a los cinco años y del 100% a los 10. Aproximadamente un tercio presentará la “forma juvenil”, con inicio de síntomas entre los tres y 17 años y una mortalidad del 30% a los cinco años y del 55% a los 10. El “subtipo adulto” es el menos frecuente, con apenas un centenar de casos recogidos en la literatura científica.
El Libmeldy ha demostrado en los ensayos que es eficaz en la forma infantil temprana si se administra antes del inicio de los síntomas y en la juvenil si se hace cuando estos apenas han empezado. “En estos casos, la cura es hasta ahora completa, aunque hace solo cuatro o cinco años que los primeros niños empezaron a recibirlo y no sabemos todavía como evolucionarán en un futuro”, precisa Del Toro.
Para Paula, que en septiembre cumplirá siete años y ahora recibe cuidados paliativos en el Hospital Niño Jesús (Madrid), el tratamiento llega tarde. “A los dos años empezó a perder la fuerza en los brazos y piernas. Ya no era capaz de sostenerse. Después empezó a hablar cada vez más lenta, cuando era una niña que no callaba”, cuenta su madre, Sandra Vaquero.
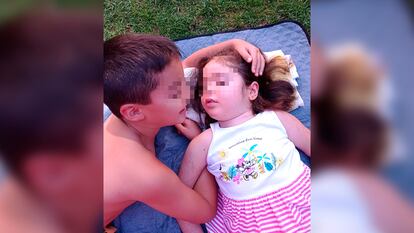
La comercialización del Libmeldy, aprobada por la Comisión Europea a finales de 2020, llegó tres años después de que Paula empezará a desarrollar síntomas claros, aunque su madre ya notaba algo desde antes. “Yo veía que en comparación con su hermano mayor, que está libre de la enfermedad, tenía menos fuerza. Pero iba bien a nivel cognitivo y era algo leve, así que los médicos no le dieron mucha importancia. Pero, de repente, todo fue a peor cuando cumplió dos años. Primero le diagnosticaron un síndrome de Dandy Walker, luego Guillain-Barré y más adelante una neuropatía desmielinizante inflamatoria. Estuvo mal diagnosticada durante más de dos años. Cuando al final supimos qué era lo que tenía, ya no cumplía los criterios para entrar en ningún ensayo clínico por lo avanzado de los síntomas”, relata la madre de Paula.
A la búsqueda del diagnóstico precoz
El caso de Paula evidencia el gran reto que tiene por delante el sistema sanitario para sacar todo el potencial al nuevo tratamiento: el diagnóstico precoz. “Los niños con síntomas ya no pueden recibirlo porque el fármaco no es eficaz. Pero ahora es casi imposible saber antes si un niño tiene la enfermedad, salvo que tenga un hermano mayor afectado y se vaya a buscar la alteración genética. Sería necesario introducir la prueba para detectar las leucodistrofias en los cribados neonatales”, señala Carmen Sever, presidenta de la Asociación Española contra la Leucodistrofia.
Ninguna comunidad autónoma incluye ahora las leucodistrofias en estas pruebas —más conocidas como del talón— que se hacen a los niños recién nacidos en España, aunque empieza a haber algunos planes pilotos. La razón es que tienen un elevado coste y, además, hasta ahora tampoco había ningún tratamiento disponible. “Pero esto está cambiando. Ya existen fármacos que curan a estos niños, así que es importante introducirlas”, añade Sever. “Nos podemos encontrar con la paradoja de que tengamos el medicamento y no nos lleguen enfermos”, ilustra por su parte Del Toro.
En España existen unas enfermedades comunes incluidas en todos los cribados neonatales fijadas por el Ministerio de Sanidad, a las que luego las comunidades pueden añadir otras. “Nos reunimos con el ministerio justo antes de la pandemia para ampliarlas, pero luego ha quedado todo parado”, lamenta Sever. Sanidad no ha informado a este diario sobre si prevé ampliar el número de enfermedades incluidas en sus recomendaciones.
Aunque la Comisión Europea autorizó hace más de un año la comercialización del Libmeldy, que todavía no está aprobado en Estados Unidos, no ha sido hasta los últimos meses cuando algunos países europeos han empezado a financiarlo en sus sistemas sanitarios. El problema principal ha sido el elevado precio que Orchard Therapeutics pide por el tratamiento. El Reino Unido rechazó en julio de 2021 las pretensiones económicas de la farmacéutica, cifradas entonces por la compañía en entre 2,5 y 3 millones de euros, alegando que era demasiado caro y no estaba demostrada su eficacia a largo plazo. Posteriormente, tras negociaciones que se han mantenido confidenciales, el país anunció en febrero un acuerdo de financiación en la sanidad pública.
Orchard Therapeutics hizo público en mayo que había alcanzado el mismo acuerdo con otros dos países, Alemania e Italia, y detalló que el precio fijado con el primero era de 2,475 millones de euros. La nota incluía una referencia a España en la que decía que la compañía preveía “acreditar” algún hospital en el país para administrar el tratamiento. Sobre la financiación pública del fármaco en España, Sanidad afirma que “está en estudio” y la compañía que no puede ofrecer detalles, ya que “las negociaciones están en marcha”. El proceso puede durar aún varios meses, durante los cuales existen mecanismos especiales para que si algún niño necesitara el fármaco pueda recibirlo.
Dos recientes informes publicados por Sanidad ponen en evidencia el impacto en las cuentas públicas que están teniendo los nuevos fármacos innovadores. En solo cinco años, el gasto público en tratamientos oncológicos ha crecido el 94% (hasta los 3.110 millones de euros) y el de los dedicados a las enfermedades raras, el 66% (hasta los 1.004 millones). “Cuando estas innovaciones llegan al mercado, es inasumible rechazarlas. Se puede negociar el precio un poco a la baja o introducir fórmulas de riesgo compartido, como el pago por resultados, como se hizo con el Zolgensma. Un abordaje mejor sería impulsar políticas bien financiadas de investigación pública en el ámbito europeo para alcanzar acuerdos de riesgo compartido con las farmacéuticas antes, cuando los medicamentos aún están en desarrollo, y no cuando este se ha completado y las empresas tienen toda la fuerza negociadora. Extender las colaboraciones público-privada durante fases más tempranas sería una mejor estrategia para contener la escalada de precios que vivimos”, concluye el profesor Juan Oliva.
Tu suscripción se está usando en otro dispositivo
¿Quieres añadir otro usuario a tu suscripción?
Si continúas leyendo en este dispositivo, no se podrá leer en el otro.
FlechaTu suscripción se está usando en otro dispositivo y solo puedes acceder a EL PAÍS desde un dispositivo a la vez.
Si quieres compartir tu cuenta, cambia tu suscripción a la modalidad Premium, así podrás añadir otro usuario. Cada uno accederá con su propia cuenta de email, lo que os permitirá personalizar vuestra experiencia en EL PAÍS.
¿Tienes una suscripción de empresa? Accede aquí para contratar más cuentas.
En el caso de no saber quién está usando tu cuenta, te recomendamos cambiar tu contraseña aquí.
Si decides continuar compartiendo tu cuenta, este mensaje se mostrará en tu dispositivo y en el de la otra persona que está usando tu cuenta de forma indefinida, afectando a tu experiencia de lectura. Puedes consultar aquí los términos y condiciones de la suscripción digital.































































