El legado de Max Perutz
M ax Perutz, premio Nobel de química en 1962 y uno de los fundadores de la biología molecular, murió el pasado miércoles, 6 de febrero. Perutz vio nacer una nueva ciencia y vivió lo suficiente -87 años- para comprobar el desarrollo extraordinario que esta iba a tener ya a finales del siglo XX. Su contribución fue decisiva, al determinar una de las primeras estructuras tridimensionales de una proteína.
¿Por qué fue esto tan importante? Si bien en el ADN está contenida la información de los seres vivos, esa información tiene que traducirse en las proteínas para ser operativa ya que éstas son las máquinas que realizan el trabajo de mantenernos vivos. Incluso el mismo ADN es leído, reparado, copiado y transferido por las proteínas. Saber cómo funcionan estas diminutas máquinas es desentrañar uno de los secretos de la vida. Pero hay más: las proteínas son las dianas de la mayor parte de los fármacos y conocer cómo son ayuda a descubrir nuevos medicamentos. Francis Crick dijo una vez: 'Si quieres comprender la función, estudia la estructura'. Eso es exactamente lo que hizo Perutz.
Las estructuras se visualizan ahora en tres dimensiones, en estaciones gráficas
Pero situémonos en 1936, cuando el joven vienés de 22 años Max Perutz, hijo de un industrial de tejidos judío, llegó al laboratorio Cavendish de la universidad de Cambridge (Reino unido) para realizar su tesis doctoral bajo la dirección de John Bernal. Se sabía, hacía tiempo, que las proteínas, a pesar de su descomunal tamaño, podían cristalizar. Si una sustancia es capaz de formar un cristal, aunque sea de menos de un milímetro, se puede, en principio, resolver su estructura atómica ya que el cristal difracta los rayos X.
Al bombardear un cristal con un fino haz de rayos X, los electrones de los átomos de la molécula cristalizada dispersan la radiación incidente en todas direcciones. Estos rayos dispersados se interfieren y anulan entre sí en la mayoría de las direcciones del espacio, pero en algunas pocas la interferencia es constructiva y se refuerzan, dando lugar al fenómeno de la difracción.
Si se pone una placa fotográfica delante, se verá que sólo se ha impresionado en unos puntos espaciados regularmente, que se llaman reflexiones. A partir de la distribución e intensidad de estos puntos, se puede reconstruir o ver la estructura molecular de la sustancia cristalizada. Pero el proceso para hacer esta reconstrucción no es sencillo porque la mitad de la información, la llamada fase de las reflexiones, no se puede medir. La cosa se complica enormemente al crecer el número de átomos de la molécula a estudiar.
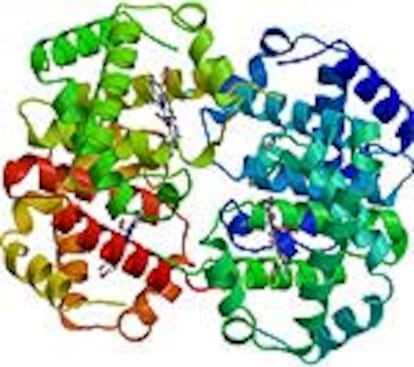
Cuando Perutz llegó a Cambridge la estructura molecular más grande que se había resuelto era la del pigmento natural ficocianina, de 58 átomos. Una proteína tiene miles de átomos. Bernal, su director, había realizado algunas imágenes de difracción de rayos X de cristales de una proteína, la pepsina, pero sin llegar a interpretarlas. El tema escogido por Perutz para su tesis fue otra proteína, la hemoglobina, el transportador de oxígeno que da color rojo a nuestra sangre. La hemoglobina tiene nada menos que 11.000 átomos.
Para hacerse una idea de la magnitud del proyecto de Perutz, es como si uno se propone escalar el Everest cuando el récord mundial de escalada fuera, tan sólo, subir por las escaleras de una casa hasta el quinto piso. Bernal y Perutz no eran unos ilusos, esperaban que la estructura de las proteínas globulares, como la hemoglobina, fuera repetitiva de forma que no fuera necesario localizar los 11.000 átomos, sólo el motivo de repetición.
Hoy se sabe que las proteínas globulares -a diferencia de las fibrosas, como la queratina del cabello- no son repetitivas. La cadena polipeptídica que las forma da vueltas y más vueltas, se contornea como un espagueti, describiendo una trayectoria impredecible, aunque algunos tramos, como las hélices, sean muy regulares. No hubo forma de evitarlo: al final se tuvo que escalar todo el Everest.
Hasta los años cincuenta Perutz no consiguió ningún resultado importante. Un modelo suyo de 1947 muestra lo lejos que estaba de la verdad. Representa la hemoglobina como una caja de sombreros, cilíndrica, con cuatro líneas circulares que son las cadenas polipeptídicas. El proyecto de Perutz había llegado a un callejón sin salida hasta que, en 1953, leyó el trabajo de un bioquímico de la universidad de Harvard que describía como al tratar la hemoglobina con paramercuriobenzoato no le pasaba nada.
A pesar de tener el mercurio enlazado, la hemoglobina seguía uniendo oxígeno. El mercurio, a diferencia de los átomos que forman las proteínas, es un átomo pesado, pesa unas diez veces más. Perutz tuvo entonces una ocurrencia genial: introdujo mercurio en los cristales de hemoglobina y tomó imágenes de difracción. Comparándolas con las de los cristales sin mercurio, vio que la intensidad de algunos puntos de difracción era diferente. Como la estructura de la hemoglobina presumiblemente no había cambiado, las diferencias se debían a la difracción añadida de los átomos de mercurio.
Así consiguió localizar estos átomos pesados primero y, con estas marcas en un paisaje confuso, empezó a trazar el camino de la proteína después. En palabras más técnicas, consiguió determinar las fases de las reflexiones o puntos de difracción. Pero se necesitaban más marcas para completarlo -más derivados de hemoglobina con otros átomos pesados-, y aún tardaron en llegar. El primer mapa de la hemoglobina interpretable no se consiguió hasta 1959. Habían pasado 23 años.
En el banco de datos de proteínas están depositadas, al día de hoy, 17.000 estructuras y su número se incrementa exponencialmente. Perutz desbrozó una vía que no ha parado de crecer. El proceso de determinación de estructuras tridimensionales de proteínas se ha acelerado considerablemente. Ya no se necesitan 20 años de esfuerzo para resolver una estructura y, hoy día, se pueden abordar estructuras muy complejas como el ribosoma, que contiene docenas de proteínas y varias moléculas de ARN.
¿Qué es lo que ha cambiado y lo que ha permanecido del método de Perutz? En primer lugar ha cambiado la disponibilidad de proteína. Antes, sólo las proteínas mayoritarias o fáciles de extraer y purificar -como la hemoglobina- se podían cristalizar. Hoy, las técnicas del ADN recombinante -clonaje, para entendernos- permiten disponer de cualquier proteína, por minoritaria que sea en la célula, si disponemos del gen que la codifica. La secuenciación del genoma humano y de otros organismos ha abierto la puerta a determinar masivamente todas las proteínas codificadas en un genoma.
Por otra parte, la cristalización no ha cambiado mucho. Se sigue haciendo de la misma forma, un poco artesanal, y bastantes proteínas se niegan a cristalizar sin que sepamos como superar este escollo. Los datos de difracción de rayos X ya no se recogen con pequeños generadores de rayos X propios. Ahora se usan sincrotrones, enormes aceleradores de electrones que producen radiación X de alta intensidad y sintonizable.
Los nuevos detectores de rayos X digitales han suprimido los viejos clichés fotográficos y la informática ha acelerado el tratamiento de los datos y los cálculos. Las estructuras se visualizan ahora en tres dimensiones, en estaciones gráficas, y los modelos de madera o varillas metálicas han pasado a los museos. Pero, para resolver las estructuras se sigue utilizando el método de Perutz. Con algunas variaciones, la idea básica sigue siendo la misma: introducir en la proteína átomos más pesados que el carbono, el nitrógeno, el oxígeno, el hidrógeno o el azufre que las componen. Un legado extraordinario de un científico de tenacidad fuera de serie.
Miquel Coll es profesor de investigación del CSIC

Tu suscripción se está usando en otro dispositivo
¿Quieres añadir otro usuario a tu suscripción?
Si continúas leyendo en este dispositivo, no se podrá leer en el otro.
FlechaTu suscripción se está usando en otro dispositivo y solo puedes acceder a EL PAÍS desde un dispositivo a la vez.
Si quieres compartir tu cuenta, cambia tu suscripción a la modalidad Premium, así podrás añadir otro usuario. Cada uno accederá con su propia cuenta de email, lo que os permitirá personalizar vuestra experiencia en EL PAÍS.
¿Tienes una suscripción de empresa? Accede aquí para contratar más cuentas.
En el caso de no saber quién está usando tu cuenta, te recomendamos cambiar tu contraseña aquí.
Si decides continuar compartiendo tu cuenta, este mensaje se mostrará en tu dispositivo y en el de la otra persona que está usando tu cuenta de forma indefinida, afectando a tu experiencia de lectura. Puedes consultar aquí los términos y condiciones de la suscripción digital.




























































